Section Contents
Biological background
The early development of X-ray crystallography (Ameh 2019Ameh, E. (2019), ‘A review of basic crystallography and x-ray diffraction applications’, The International Journal of Advanced Manufacturing Technology 105(7), 3289–3302, doi.org/10.1007/s00170-019-04508-1, Blundell et al. 1971Blundell, T., Dodson, G., Dodson, E., Hodgkin, D. & Vijayan, M. (1971), X-ray analysis and the structure of insulin, in ‘Proceedings of the 1970 Laurentian Hormone Conference’, Elsevier, pp. 1–40, doi.org/10.1016/B978-0-12-571127-2.50025-0.) as a means of determining the structure of proteins meant that most work in structural biology in the 20th century tried to explain the functions of proteins by considering them as static structures (Karplus & McCammon 1986Karplus, M. & McCammon, J. A. (1986), ‘The dynamics of proteins’, Scientific American 254(4), 42–51.). However, recent decades have seen a shift towards a greater appreciation for the role that dynamics plays in determining protein function (Zhao 2013Zhao, Q. (2013), ‘Nature of protein dynamics and thermodynamics’, Reviews in Theoretical Science 1(1), 83–101, doi.org/10.1166/rits.2013.1005., Doniach & Eastman 1999Doniach, S. & Eastman, P. (1999), ‘Protein dynamics simulations from nanoseconds to microseconds’, Current Opinion in Structural Biology 9(2), 157–163, doi.org/10.1016/S0959-440X(99)80022-0.), largely thanks to new techniques such as protein NMR (Sekhar & Kay 2019Sekhar, A. & Kay, L. E. (2019), ‘An nmr view of protein dynamics in health and disease’, Annual review of biophysics 48, 297–319, doi.org/10.1146/annurev-biophys-052118-115647., Ishima & Torchia 2000Ishima, R. & Torchia, D. A. (2000), ‘Protein dynamics from nmr’, Nature structural biology 7(9), 740–743, doi.org/10.1038/78963.) and XFELs (Spence 2017Spence, J. (2017), ‘Xfels for structure and dynamics in biology’, IUCrJ 4(4), 322–339, doi.org/10.1107/S2052252517005760., Johansson et al. 2017Johansson, L. C., Stauch, B., Ishchenko, A. & Cherezov, V. (2017), ‘A bright future for serial femtosecond crystallography with xfels’, Trends in biochemical sciences 42(9), 749–762, doi.org/10.1016/j.tibs.2017.06.007.). Indeed, much has been made recently of intrinsically disordered proteins which do not fold into a stable structure at all, yet exhibit biological functions (Wright & Dyson 2015Wright, P. E. & Dyson, H. J. (2015), ‘Intrinsically disordered proteins in cellular signalling and regulation’, Nature reviews Molecular cell biology 16(1), 18–29, doi.org/10.1038/nrm3920., Oldfield & Dunker 2014Oldfield, C. J. & Dunker, A. K. (2014), ‘Intrinsically disordered proteins and intrinsically disordered protein regions’, Annual review of biochemistry 83, 553–584, doi.org/10.1146/annurev-biochem-072711-164947.). Therefore, having accurate models of protein dynamics will be essential for almost all future work in biology, but especially drug design (Lee et al. 2019Lee, Y., Lazim, R., Macalino, S. J. Y. & Choi, S. (2019), ‘Importance of protein dynamics in the structure based drug discovery of class ag protein-coupled receptors (gpcrs)’, Current opinion in structural biology 55, 147–153, doi.org/10.1016/j.sbi.2019.03.015., Mortier et al. 2015Mortier, J., Rakers, C., Bermudez, M., Murgueitio, M. S., Riniker, S. & Wolber, G. (2015), ‘The impact of molecular dynamics on drug design: applications for the characterization of ligand–macromolecule complexes’, Drug Discovery Today 20(6), 686–702, ., Zhao & Caflisch 2015Zhao, H. & Caflisch, A. (2015), ‘Molecular dynamics in drug design’, European journal of medicinal chemistry 91, 4–14, doi.org/10.1016/j.ejmech.2014.08.004., Peng 2009Peng, J. W. (2009), ‘Communication breakdown: protein dynamics and drug design’, Structure 17(3), 319–320, doi.org/10.1016/j.str.2009.02.004.) and understanding diseases caused by misfolding proteins, such as Alzheimer’s and Parkinson’s (Selkoe 2004Selkoe, D. J. (2004), ‘Cell biology of protein misfolding: the examples of alzheimer’s and parkinson’s diseases’, Nature cell biology 6(11), 1054–1061, doi.org/10.1038/ncb1104-1054.).
The advances of AlphaFold2 (Jumper et al. 2020)Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Tunyasuvunakool, K., Ronneberger, O., Bates, R., Zidek, A., Bridgland, A. et al. (2020), ‘High accuracy protein structure prediction using deep learning’, Fourteenth Critical Assessment of Techniques for Protein Structure Prediction (Abstract Book) 22, 24. have shown that our future understanding of biology and the mechanisms of disease will be increasingly understood at the structural level (AlQuraishi 2021) AlQuraishi, M. (2021), ‘Machine learning in protein structure prediction’, Current Opinion in Chemical Biology 65, 1–8, doi.org/10.1016/j.cbpa.2021.04.005.. Therefore, developing new ”neuralised” approaches to understand the dynamics of proteins will be of fundamental importance to solve problems at the frontiers of medicine and drug design. Indeed, the fact that a limitation of AlphaFold2 seems to be its ability to predict NMR solved structures (Jumper et al. 2020) suggests there is still much work to be done.
Machine learning background
Graph Neural Networks (GNNs) (Battaglia et al. 2018Battaglia, P. W., Hamrick, J. B., Bapst, V., Sanchez-Gonzalez, A., Zambaldi, V., Malinowski, M., Tacchetti, A., Raposo, D., Santoro, A., Faulkner, R. et al. (2018), ‘Relational inductive biases, deep learning, and graph networks’, arXiv preprint arXiv:1806.01261 ., Monti et al. 2017Monti, F., Boscaini, D., Masci, J., Rodola, E., Svoboda, J. & Bronstein, M. M. (2017), Geometric deep learning on graphs and manifolds using mixture model cnns, in ‘Proceedings of the IEEE conference on computer vision and pattern recognition’, pp. 5115–5124.), themselves a subset of Geometric Deep Learning (Bronstein et al. 2017Bronstein, M. M., Bruna, J., LeCun, Y., Szlam, A. & Vandergheynst, P. (2017), ‘Geometric deep learning: going beyond euclidean data’, IEEE Signal Processing Magazine 34(4), 18–42, doi.org/10.1109/MSP.2017.2693418), have been used to learn complex physics before (Sanchez-Gonzalez et al. 2020Sanchez-Gonzalez, A., Godwin, J., Pfaff, T., Ying, R., Leskovec, J. & Battaglia, P. (2020), Learning to simulate complex physics with graph networks, in ‘International Conference on Machine Learning’, PMLR, pp. 8459–8468.). However, in comparison to proteins, these have all been done on relatively homogeneous materials and little is known about the number of parameters and expressivity required to learn the dynamics of whole proteins (and protein complexes) using neural network based approaches.
There has also been much recent work in developing deep learning architectures that are equivariant to rigid motions in Euclidean space (Satorras, Hoogeboom & Welling 2021Satorras, V. G., Hoogeboom, E., Fuchs, F. B., Posner, I. & Welling, M. (2021), ‘E(n) equivariant normalizing flows for molecule generation in 3d’, arXiv preprint arXiv:2105.09016 ., Fuchs et al. 2020Fuchs, F. B., Worrall, D. E., Fischer, V. & Welling, M. (2020), ‘Se (3)-transformers: 3d roto-translation equivariant attention networks’, arXiv preprint arXiv:2006.10503 .) and able to learn higher-order interactions in graph structured data (Bodnar et al. 2021Bodnar, C., Frasca, F., Wang, Y. G., Otter, N., Montu ́far, G., Lio, P. & Bronstein, M. (2021), ‘Weisfeiler and lehman go topological: Message passing simplicial networks’, arXiv preprint arXiv:2103.03212.) that have yet to gain much attention in the domain of structural biology, let alone protein dynamics.
Related work
This work was motivated in large part by Greener & Jones (2021)
Greener, J. G. & Jones, D. T. (2021),
‘Differentiable molecular simulation can learn all the parameters in a coarse-grained force field for proteins’,
bioRxiv
doi:10.1101/2021.02.05.429941
which proposed a novel Differentiable Molecular Simulation (DMS) for proteins. Here, the authors showed that all the parameters in a conventional, course-grained force-field could be learnt using automatic differentiation (AD). Interestingly, their loss function was simply that the overall protein structure was maintained after being passed through the learnt dynamics simulator with random initial velocities assigned to all the atoms. In other words, if the Root Mean Squared Deviation (RMSD) of all the atoms in the proteins was sufficiently low after N time steps, then the model can be considered to have learnt a form of dynamics that is analogous to that seen in native proteins.
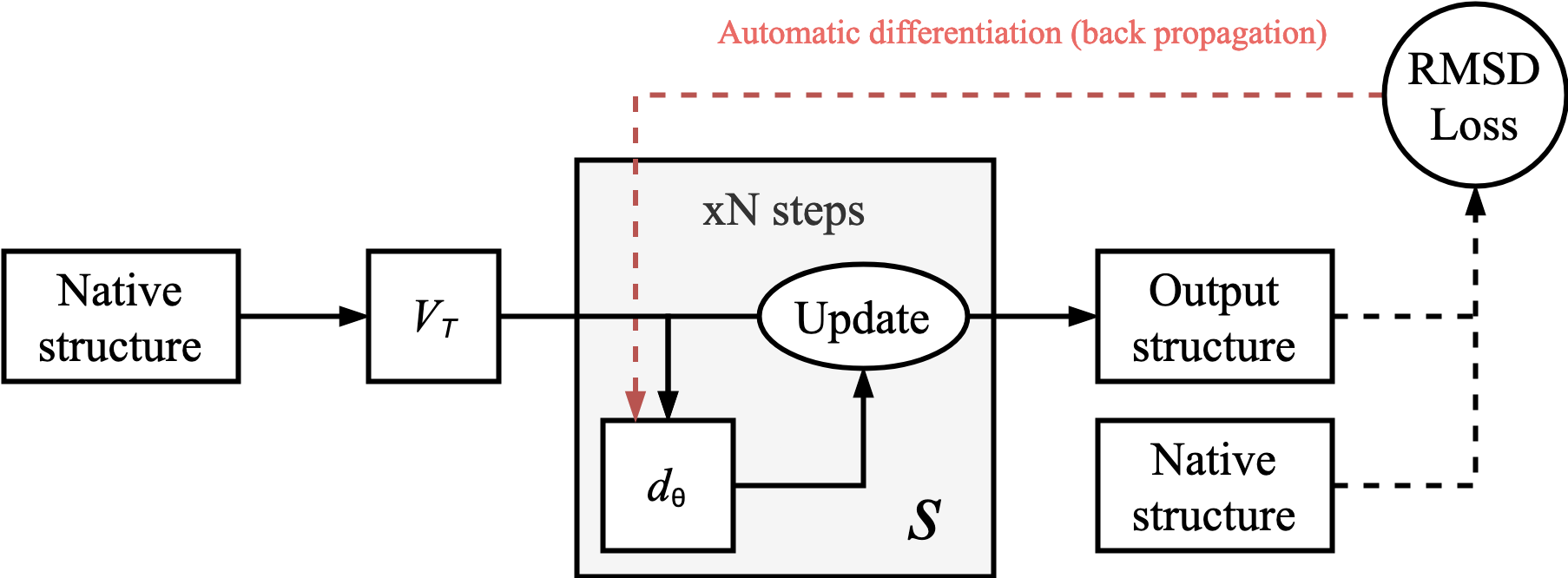
A generalised overview of the approach outlined in Greener & Jones (2021)
Greener, J. G. & Jones, D. T. (2021),
‘Differentiable molecular simulation can learn all the parameters in a coarse-grained force field for proteins’,
bioRxiv
doi:10.1101/2021.02.05.429941 is provided in Figure 1. The learned dynamics model, dθ, can be a conventional force-field (with differentiable parameters) or a neural network. Greener & Jones (2021)
Greener, J. G. & Jones, D. T. (2021),
‘Differentiable molecular simulation can learn all the parameters in a coarse-grained force field for proteins’,
bioRxiv
doi:10.1101/2021.02.05.429941 demonstrated that all the parameters needed in a coursegrained force field could be learnt in this manner by assigning individual energy potentials for every possible atom-atom interaction that is split into 140 bins. The forces were calculated by taking the gradients between adjacent bins whilst the atoms were a certain distance apart. Due to the nature of AD requiring intermediate values of the equations to be stored, the memory requirements of the simulation scales linearly with the number of time steps and is thus the limiting factor in DMS.
However, there were significant shortcomings with this approach that need to be addressed. Firstly, an inter-atomic distance matrix needs to be calculated at every step in the simulation. This incurs significant processing and memory costs which were prohibitive to training on proteins greater than 100 amino acids in length or having greater than 2000 time steps. Secondly, the model assumes that every possible atom-atom interaction will share no features and thus defines individual potentials for all of them. This leads to a large number of parameters (4,000,000+) that needs to be learnt and optimised separately leading to poor generalisation. All of these factors led to significant training times (2 months on a Nvidia Tesla V100), which is out of the scope of this project. We argue that this can be reframed as a multi-task learning problem, where we can exploit the commonalities and differences in the different types of interactions seen in proteins.
The development of DMS libraries, including TorchMD (Doerr et al. 2021Doerr, S., Majewski, M., P ́erez, A., Kramer, A., Clementi, C., Noe, F., Giorgino, T. & De Fabritiis, G. (2021), ‘Torchmd: A deep learning framework for molecular simulations’, Journal of Chemical Theory and Computation 17(4), 2355–2363.), JAX MD (Schoenholz & Cubuk 2019Schoenholz, S. S. & Cubuk, E. D. (2019), ‘Jax md: End-to-end differentiable, hardware accelerated, molecular dynamics in pure python’.), DeePMD-kit (Wang et al. 2018Wang, H., Zhang, L., Han, J. & Weinan, E. (2018), ‘Deepmd-kit: A deep learning package for many body potential energy representation and molecular dynamics’, Computer Physics Communications 228, 178–184, doi.org/10.1016/j.cpc.2018.03.016.), SchNetPack (Schutt et al. 2018Schutt, K., Kessel, P., Gastegger, M., Nicoli, K., Tkatchenko, A. & Muller, K.-R. (2018), ‘Schnetpack: A deep learning toolbox for atomistic systems’, Journal of chemical theory and computation 15(1), 448– 455, /doi.org/10.1021/acs.jctc.8b00908.) and DiffTaichi (Hu et al. 2019Hu, Y., Anderson, L., Li, T.-M., Sun, Q., Carr, N., Ragan-Kelley, J. & Durand, F. (2019), ‘Difftaichi: Differentiable programming for physical simulation’, arXiv preprint arXiv:1910.00935 .), will hopefully spawn much more work into learning protein dynamics using DMS.
Aims and motivations
Thus, the aims of this work are 4 fold:- Create a DMS system that can learn the dynamics of a single protein domain by only requiring a small amount of training data. This is so that the techniques developed here can be transferred to other problems where there will likely be less training data available (e.g protein-protein and protein-drug interactions).
- Reducing the number of parameters that need to be learnt in the force field by leveraging the high generalising power and expressivity of modern deep learning architectures.
- Maintaining interpretability in the learnt dynamics by integrating our knowledge of protein dynamics into our machine learning models.
- Reduce the large memory requirements seen in Greener & Jones (2021)
Greener, J. G. & Jones, D. T. (2021),
‘Differentiable molecular simulation can learn all the parameters in a coarse-grained force field for proteins’,
bioRxiv
doi:10.1101/2021.02.05.429941 by using the KeOps (Charlier et al. 2021Charlier, B., Feydy, J., Glaun`es, J., Collin, F.-D. & Durif, G. (2021), ‘Kernel operations on the gpu, with autodiff, without memory overflows’, Journal of Machine Learning Research 22(74), 1–6.) library to calculate distance matrices symbolically.